
The RAS oncogenes are the most frequently mutated class of oncogenes in human cancers, and this has prompted a search for Ras inhibitors to effectively treat tumors with these mutations. Despite intensive efforts, however, none has materialized clinically because K-Ras is proving to be a very vexing target, according to Channing J. Der, PhD, of the University of North Carolina at Chapel Hill, who gave an invited lecture on the topic at the 2012 Gastrointestinal Cancers Symposium.1,2
The search for K-Ras inhibitors, he said, is like the hunt for the Holy Grail in cancer research. “It has not been easy, though lessons have been learned from our research, some of them bitter and disappointing,” he noted.
“Targeting effector signaling is our best hope. However, a cocktail of inhibitors hitting pathways at multiple points will be needed, and this cocktail will be different, depending upon the Ras isoform, the particular mutation, and the cancer type. This is much more complicated than we had previously envisioned,” Dr. Der said.
Lessons Learned about K-Ras
 Four main “lessons” have emerged from almost 3 decades of research:
Four main “lessons” have emerged from almost 3 decades of research:
- The three RAS genes (KRAS, NRAS, and HRAS) are not identical in function. KRAS is the most important isoform mutated in colorectal cancer.
- The K-Ras protein is considered “undruggable.” Current efforts target K-Ras indirectly.
- Signal transduction pathways are not simple linear unidirectional pathways, but are complex, dynamic networks that are difficult to fully comprehend.
- The 40% of KRAS mutant colorectal cancers are genetically and biologically heterogeneous. One therapeutic approach alone will not work.
“In colorectal cancer, many mutations are identified (approximately 80 per tumor), but most are harmless, and the development of cancer is driven by about 15. KRAS is the key oncogene and the most attractive for targeting. But while 40% of tumors have KRAS mutations, they also share—at a much lower frequency—other mutations, and as a consequence are genetically heterogeneous,” Dr. Der noted.
Targeting all the necessary mutation combinations within one tumor will be problematic. And should this be accomplished—that is, should the KRAS defect be “corrected”—will this alone have a positive clinical impact? While this remains unknown, it is clear that colorectal cancer cells are “addicted” to K-Ras—ie, they require its presence, making it a validated target. Refining the target and developing drugs for it should have significant therapeutic consequences, he predicted.
Future Therapeutic Options
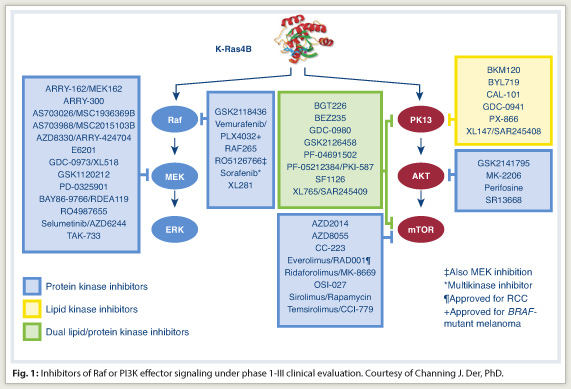 “Ideally, what needs to be done is to target K-Ras directly, but we have not succeeded in this. As a result, there is a perception that Ras is an undruggable target,” he said. For complex biochemical reasons, it is essentially impossible to develop a small-molecule antagonist that will block GTP binding, which is critical for Ras function.
“Ideally, what needs to be done is to target K-Ras directly, but we have not succeeded in this. As a result, there is a perception that Ras is an undruggable target,” he said. For complex biochemical reasons, it is essentially impossible to develop a small-molecule antagonist that will block GTP binding, which is critical for Ras function.
Ras is a signaling molecule that stimulates a cascade of cytoplastic signaling pathways. Activated Ras binds preferentially to a spectrum of functionally diverse downstream effectors. These include the protein kinases Raf, MEK, and ERK of the MAPK cascade, and the lipid kinase PI3K, which activates the AKT and mTOR protein kinases. At least three other functionally distinct classes of Ras effectors exist, with validated roles in Ras transformation. Significant evidence now suggests the potential importance of these less studied effectors for anti-Ras drug discovery, Dr. Der said.
“Most of the effort in developing Ras inhibitors is focusing on the inhibition of downstream effector signaling,” he said. “If we block these pathways successfully, we should be able to cripple Ras.”
Approximately 40 small-molecule inhibitors of different components of these effector pathways are now in clinical trials (Fig. 1). However, targeting Ras effector signaling is not simple . It is now known that Raf inhibition and MEK inhibition do not produce the same effect and that Ras does not rely exclusively on the MAP kinase pathway, accentuating the concept that “multiple effectors are necessary for Ras-dependent cancer growth,” Dr. Der noted.
Further Complications
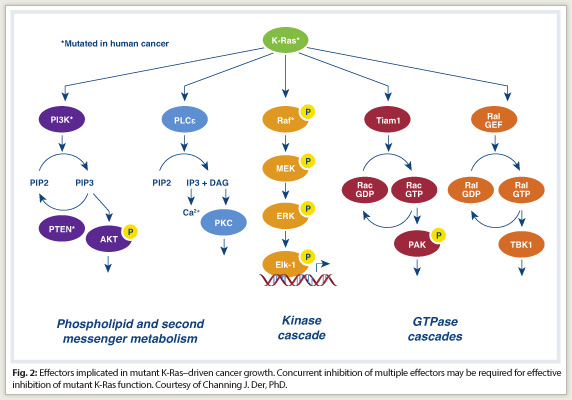 In addition, mutant KRAS does not correlate with ERK activation, and inhibition of B-raf in colorectal cancer does not produce the same clinical effect as it does in melanoma. “This suggests that the same genetic mutation in two different cancer types is not susceptible to the same treatment approach,” he said.
In addition, mutant KRAS does not correlate with ERK activation, and inhibition of B-raf in colorectal cancer does not produce the same clinical effect as it does in melanoma. “This suggests that the same genetic mutation in two different cancer types is not susceptible to the same treatment approach,” he said.
Furthermore, MEK inhibitors do not reduce the growth of KRAS mutant colorectal cancer cells. “This was surprising and disappointing,” he said. “We found that most KRAS-mutant cell lines were refractory to growth inhibition with MEK inhibitors, despite the fact that MEK inhibition did block the activation of this pathway,” Dr. Der said.
Compensatory mechanisms take over, he explained, saying “This is not the only pathway that Ras depends on for initiating oncogenic consequences. Concurrent inhibition of two or more pathways will be needed to effectively ablate oncogenic KRAS-driven growth (Fig. 2).”
Additionally, the core effector signaling pathways are probably different among the cancer types. All KRAS mutants “are not created equal”—ie, the frequency of specific mutations varies across cancer types—and clinical response varies according to the specific mutation.
All things considered, Dr. Der predicted that inhibitors of effector signaling will ultimately be effective against KRAS-mutant cancers, but therapeutic “cocktails” unique to the Ras isoform, the particular RAS mutant, and the cancer type will be needed for full therapeutic efficacy. ■
Disclosure: Dr. Der reported no potential conflicts of interest.
References
1. Der CJ: Targeting KRAS for the treatment of gastrointestinal cancers: Mission impossible? Invited lecture. 2012 Gastrointestinal Cancers Symposium. Presented January 21, 2012.
2. Cox AD, Der CJ: Ras history: The saga continues. Small GTPases 1:2-27, 2010.