



The FDA’s approval process ensures that brand-name and generic drugs meet rigorous standards for safety and efficacy. Postapproval safety surveillance monitoring along with drug product quality testing enables the FDA to ensure drug product safety and quality.— Kathleen Uhl, MD, John R. Peters, MD, and Howard Chazin, MD, MBA
Tweet this quote
The U.S. Food and Drug Administration’s (FDA) generic drug program has substantially increased the availability of affordable, high-quality drugs in the United States. It is arguably the only really effective health-care cost–containment program. The more than 10,000 generic drugs currently approved saved the health-care system approximately $1.46 trillion in the past 10 years.1
Generic drugs currently make up nearly 9 of 10 prescriptions dispensed in the United States. Dispensing of oncology generic drugs is expected to mirror this rate by 2020.2 Clinicians, patients, researchers, and others sometimes ask questions about the quality, safety, and efficacy of generic drugs, as evidenced by the report by Yang and colleagues in The Lancet Oncology and summarized in this issue of The ASCO Post.3
Determining Equivalence and Bioequivalence
Generic drug manufacturers must demonstrate that their drug product is pharmaceutically equivalent and bioequivalent to the brand-name drug (also known as the Reference Listed Drug) to receive FDA approval. These analyses allow the FDA to determine that the generic drug product is as safe and effective as the Reference Listed Drug.
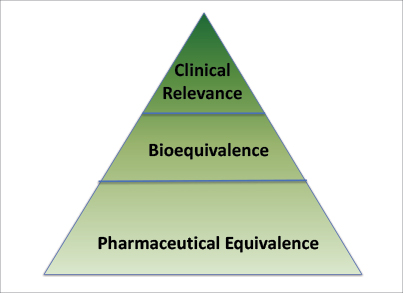
Fig. 1: Foundation of Generic Drug Review Assesement.
Pharmaceutical equivalence is the foundation for generic equivalence (Fig. 1) and means that the generic drug is the same as the brand-name drug (Reference Listed Drug) in terms of the following:
- Active ingredient;
- Dosage form (eg, tablet with tablet, not capsule with tablet);
- Strength (eg, 40 mg Reference Listed Drug with 40 mg generic);
- Route of administration;
- Manufactured under the FDA’s strict standards of Good Manufacturing Practice regulations;
- Conditions of use.
Generic drug manufacturers must also demonstrate that their drug product is bioequivalent to the Reference Listed Drug. Bioequivalence means that the rate and extent of absorption of the generic does not show a significant difference from the rate and extent of absorption of the Reference Listed Drug. Bioequivalence assures the FDA that the generic will perform physiologically in the same way as the Reference Listed Drug. A pharmacokinetic study in healthy human subjects is typically conducted to demonstrate bioequivalence, but pharmacokinetic studies for oncology drugs and other types of cytotoxic or genotoxic drugs are usually conducted in patients.
Bioequivalent analysis includes a robust statistical evaluation of pharmacokinetic data, maximum concentration (Cmax) and area under the concentration-time curve (AUC), comparing the generic with the Reference Listed Drug. Statistical analysis requires that the ratio (generic: Reference Listed Drug) of these measures remains strictly within a 90% confidence interval, with a range of 0.80 to 1.25. A retrospective study of more than 2,000 generic drugs demonstrated that, using these measures, the average difference in Cmax and AUC between generic and innovator products was 4.35% and 3.56%, respectively.4 The FDA has published more than 1,500 product-specific guidances to aid generic manufacturers in the study design and data analysis of bioequivalent studies.5
Finally, a more complete understanding of bioequivalence is provided by considering clinical relevance. The FDA’s analysis is done with a thorough understanding of the drug’s indication, population to be treated, chronicity of the therapy, and intended physiologic behavior of the Reference Listed Drug. This analysis is particularly important for complex dosage forms and modified-release formulations. However, generics may differ from the Reference Listed Drugs with respect to shape, scoring configuration, release mechanism, packaging, excipients, expiration date, and storage requirement and may have only minor labeling (or package insert) differences.
Regulatory, Legal, and International Aspects
The Drug Price Competition and Patent Term Restoration Act of 1984 (Hatch-Waxman Amendments) granted generic drug companies an abbreviated regulatory pathway, and brand-name drug companies market exclusivity and certain patent restorations. The Abbreviated New Drug Application (ANDA) pathway eliminated costly, redundant animal and human studies, instead leveraging the safety and efficacy data from the Reference Listed Drug’s application (New Drug Application [NDA]; Table 1). Eliminating repetitive studies and focusing on “sameness” directly resulted in an increased number of Abbreviated New Drug Applications submitted to the FDA and increased generic drug availability in the United States.

Yang et al compared oncology generic drugs globally, but it is worthwhile noting that direct comparison of generic products across different regulatory bodies is particularly challenging—and potentially not valid—due to differing laws and regulations in each region. Additionally, taking into account the large market share of generic oncology drug products vs brand-name oncology drug products dispensed, the proportion of Class I recalls for generic to brand-name drug products mentioned by Yang et al (13 generic to 4 brand) is proportional.3 All FDA-approved drug products—whether generic or brand name—are held to the same standards for safety, efficacy, and quality.
Drug Safety Efforts
The FDA’s Center for Drug Evaluation and Research (CDER) maintains a rigorous pre- and postmarket process for evaluating drug safety. Approval decisions are based on a comprehensive assessment of the benefits of the drug and its known and potential risks. Reference Listed Drug and generic drug safety are monitored across the product life cycle—from premarket development to postmarket use. A recent report highlighted the broad scope of the Center’s drug safety efforts and emphasized the critical advantages of multidisciplinary collaborations and partnerships.6 The FDA encourages consumers and health-care professionals to report adverse side effects to the FDA’s MedWatch.7
The CDER’s Office of Generic Drugs Clinical Safety and Surveillance Staff facilitates and integrates broad collaborative surveillance projects with many FDA disciplines and establishes partnerships that further the CDER’s commitment to broad, integrated surveillance of generic drug quality and therapeutic equivalence. An interdisciplinary team of physicians, pharmacists, epidemiologists, chemists, and other scientific experts track and evaluate reports relating to generic drug product quality, adverse events, or differing therapeutic effects from the brand-name drug.
The FDA investigates reports of potential therapeutic inequivalence thoroughly and tests raw ingredients and finished drug products for product quality.8 Investigations may lead to changes in how a brand-name drug and/or its generic counterparts are manufactured, packaged, or administered.
Closing Comments
The FDA’s approval process ensures that brand-name and generic drugs meet rigorous standards for safety and efficacy. Postapproval safety surveillance monitoring along with drug product quality testing enables the FDA to ensure drug product safety and quality. These activities are also critical to evaluating and ensuring that affordable, high-quality, safe, and effective drugs are available to the American public. ■
Disclosure: Drs. Uhl, Peters, and Chazin are all employees of the FDA.
References
1. Generic Pharmaceutical Association 2016 Generic Drug Savings and Access in the United States Report. Available at http://www.gphaonline.org/media/generic-drug-savings-2016/index.html. Accessed November 30, 2016.
2. Allied Market Research: World oncology/cancer drugs market—opportunities and forecasts, 2013-2020. Available at http://www.alliedmarketresearch.com/oncology-cancer-drugs-market. Accessed November 30, 2016.
5. U.S. Food & Drug Administration: Product-Specific Recommendations for Generic Drug Development. Available at http://www.fda.gov/drugs/guidancecomplianceregulatoryinformation/guidances/ucm075207.htm. Accessed November 30, 2016.
6. U.S. Food and Drug Administration: Center for Drug Evaluation and Research 2015-2016 Drug Safety Priorities: Initiatives and Innovation. Available at http://www.fda.gov/downloads/Drugs/DrugSafety/UCM523486.pdf. Accessed November 30, 2016.
7. U.S. Food and Drug Administration: MedWatch Online Voluntary Reporting Form. Available at https://www.accessdata.fda.gov/scripts/medwatch/index.cfm?action=reporting.home. Accessed November 30, 2016.
8. U.S. Food and Drug Administration: Drug Quality Sampling and Testing Programs. Available at http://www.fda.gov/Drugs/ScienceResearch/ucm407277.htm. Accessed November 30, 2016.
