
We know from chaos theory that even if you had a perfect model of the world, you’d need infinite precision in order to predict future events.
—Nassim Nicholas Taleb
The term “precision oncology” is used to describe diverse strategies in cancer medicine ranging from the use of targeted therapies generally to using data from next-generation sequencing to select therapy for a person independent of cancer type. There is widespread, even extraordinary enthusiasm for precision oncology. But is this enthusiasm justified by biologic and conceptual considerations and by data from clinical trials?


Precision therapy requires precision data. Unfortunately, we are not there yet.— Vinay Prasad, MD, MPH, and Robert Peter Gale MD, PhD, DSc(hc), FACP, FRSM
Tweet this quote
Here we describe the challenges of defining precision oncology and applying this concept to treating cancer. Consideration of the complex biology of cancers leads us to suggest caution in estimating the impact of precision oncology in cancer therapy.
Evolving Definition
Exactly which activities and actions fall under the domain of precision oncology is unclear. Collins and Varmus argue that blood-typing, targeted therapies, and even immune therapy are examples of precision oncology.1 This definition seems overly broad. Others use the term more narrowly—for example, to refer to the use of next-generation sequencing of cancer tissues to guide targeted therapy. Ironically, imprecise use of the word “precision” constitutes jargon, as defined by the Oxford English Dictionary: “special words or expressions used by a profession or group that are difficult for others to understand.”
To understand what precision oncology means in current contexts, we analyzed the use of this term in the biomedical literature by searching Google Scholar for “precision oncology” over 3 intervals and by classifying 50 articles. Results are displayed in Fig. 1.
Over these intervals, the use of the term has changed. In the earliest interval, “precision oncology” predominantly described targeted therapies such as vascular endothelial growth factor (VEGF) inhibitors or BCR/ABL1 inhibitors, including bevacizumab (Avastin) and imatinib. However, this use has largely ceased.
Subsequently, precision oncology was used to describe the selection of therapies based on data from analyses of biomarkers. Examples include the use of crizotinib (Xalkori) in persons with lung cancer with EML4-ALK rearrangements or adjuvant chemotherapy guided by the results of genomic testing such as the Oncotype DX panel in women with breast cancer.
By 2016, things changed again. Currently, precision oncology principally refers to using data from next-generation sequencing to guide therapies, a definition that was used in 15 of 21 articles we reviewed, all published since January 1, 2016.
The most paradigm-shifting definition of “precision oncology” is this final one: directing therapy independent of cancer type as currently defined (based on anatomy and histology) and instead by mutation. For instance, persons with BRAF V600E mutations would be treated similarly with a BRAF inhibitor regardless of whether they have acute myeloid leukemia or breast cancer.
Will Precision Oncology Work?
The practical case against precision oncology is that it is unlikely to benefit most people with cancer. Data from next-generation sequencing of persons with advanced cancers indicate fewer than 10% have actionable mutations.2,3
The only randomized trial of precision medicine, the SHIVA trial, tested whether genetic analyses and pathway-directed therapy produce better outcomes than investigator choice and found no progression-free survival difference between these strategies.4 This was a phase II study without blinding, and although these data do not disprove the hypothesis that a mutation profile–based strategy for selecting therapy of advanced cancers might work, it is thus far the only credible study we have.
The biologic case against precision oncology is that it contrasts with our modern understanding of cancer. The mutation landscape of most common cancers is highly complex. The least mutated cancers have on average 0.28 mutations per megabase (a term used to measure the numbers of base pairs in a stretch of DNA), and the most mutated have 8.15 mutations per megabase.5 Reports of mutation analyses by whole-exome sequencing of a moderately mutated disease like pancreatic cancer (mean, 2.64 mutations per megabase) indicate very few consistently mutated genes (and even common mutations such as NRAS occur only in some cases of pancreatic cancer, and even occur in normal tissue without pancreatic cancer).6
Although many of these mutations are passenger mutations (conferring no survival advantage on the clone), even driver mutations (which propel cancer progression) occur at low prevalence.7 There are, of course, exceptions such as NPM1 and FLT3 in acute myeloid leukemia and MYC in lymphomas, but this situation is less common in most solid neoplasms. Moreover, mutations such as NPM1 and MYC are not currently actionable—we have no drug to halt their activity.
Unique amplifications and copy number gains of oncogenes also occur at low prevalence.7 Because most people with pancreatic cancer have unique mutation fingerprints, researchers note a substantial diversity of mechanisms involved in pancreatic cancer progression.7 In other words, many roads lead to Rome.
Precision oncology is seductive and appealing.… But like all seductive notions, we must separate evidence supporting a claim from our desire for the claim to be true. In the case of precision medicine, we need clear definitions and criteria for success or failure. We are hoping for success, but there remains much imprecision to this strategy.— Vinay Prasad, MD, MPH, and Robert Peter Gale, MD, PhD, DSc(hc), FACP, FRSM
Tweet this quote
Precision oncology is based on the notion that in the midst of a shattered genome, identifying and reversing one target, a driver mutation, will dramatically halt the aberrant phenotype that characterizes the cancer and ideally restore normalcy. It is as if a city planner studied Rome’s rush hour traffic, noting many congested freeways, standstills on crossroads, bloated on-ramps, and crawling buses, and thought, “if only I could build one bridge,” the traffic problem would be solved. Even at the outset, this seems implausible save for rare cancers such as chronic myeloid leukemia, where one mutation, BCR/ABL1, is necessary and sufficient to cause the disease in most instances.
Alternatively, some experts think targeting multiple pathways might solve the problem—a series of bridges. The simple reality is that drug toxicities are often additive and unpredictable, and the history of targeted therapy has failed to validate promising combinations in many settings, even when each component drug is effective and safe.
Moreover, with the rise of commercial tests, one worries about data consistency and reproducibility. Recent data compared results of genetic variant testing for inherited cancer susceptibility between laboratories. Disconcertingly, the authors found that 11% of people tested had a variant that was reported inconsistently—not pathologic in some analyses, pathologic in others. A comparison of two commercially available next-generation sequencing platforms revealed rampant disagreements in mutation and recommended drugs for the same nine patients with relapsed/refractory cancer whose biopsy was sent for both tests.8
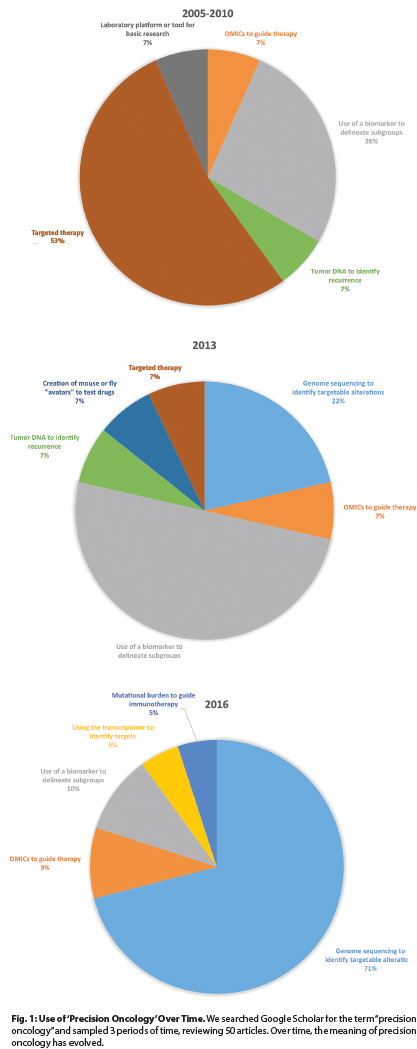
Fig. 1: Use of ‘Precision Oncology’ Over Time. We searched Google Scholar for the term “precision oncology” and sampled 3 periods of time, reviewing 50 articles. Over time, the meaning of precision oncology has evolved.
One wonders how a larger analysis would fare. Given cancer heterogeneity and limited biopsy samples, temporal clonal selection, and laboratory errors, many people would receive different therapies if the same tissue samples were tested in different laboratories. It is also important to recall most people with cancer die of one or more metastases rather than cancer at the primary site. However, in most instances, it is the primary site that is being tested for actionable mutations.
Precision therapy requires precision data. Unfortunately, we are not there yet.
When one considers the biologic and practical challenges of precision oncology—that we are performing an imprecise sequencing effort that will likely find many alterations in a damaged genome; that we may infrequently encounter an alteration targeted by a drug we already have; that we may give a drug hoping there are limited alternative pathways available for upregulation; that, all the while, we hope for a sustained and meaningful response—the entire proposition seems uncertain at best.
Precision oncology is seductive and appealing. Who wouldn’t want to use a cancer drug that will be effective in people for whom other therapy options fail? But like all seductive notions, we must separate evidence supporting a claim from our desire for the claim to be true. In the case of precision medicine, we need clear definitions and criteria for success or failure. We are hoping for success, but there remains much imprecision to this strategy. ■
Disclosure: Dr. Prasad reported no potential conflicts of interest. Dr. Gale is a part-time employee of Celgene Corp.
References
1. Collins FS, Varmus H: A new initiative on precision medicine. N Engl J Med 372:793-795, 2015.
2. ECOG-ACRIN Cancer Research Group: Executive Summary: Interim Analysis of the NCI-MATCH Trial. Available at https://dctd.cancer.gov/majorinitiatives/NCI-MATCH_Interim_Analysis_Executive_Summary.pdf. Accessed January 6, 2017.
6. Gale RP: Recent progress and concepts in pancreatic cancer. The ASCO Post. November 25, 2016. Available at www.ascopost.com. Accessed January 11, 2017.