
 Thrombocytosis is defined as a platelet count greater than 400 × 109/L. In routine clinical practice, thrombocytosis is much more likely to be reactive (> 80% of cases) than primary. Reactive thrombocytosis is usually associated with infections, inflammation, trauma, hemolysis, metastatic cancer, the postsplenectomy state, or iron deficiency anemia. Causes of primary thrombocytosis include myeloproliferative neoplasms, myelodysplastic syndromes, myelodysplastic syndrome/myeloproliferative neoplasm overlap syndromes, and other myeloid malignancies. Congenital thrombocytosis is extremely rare and sometimes associated with germline mutations of thrombopoietin or its receptor (MPL).
Thrombocytosis is defined as a platelet count greater than 400 × 109/L. In routine clinical practice, thrombocytosis is much more likely to be reactive (> 80% of cases) than primary. Reactive thrombocytosis is usually associated with infections, inflammation, trauma, hemolysis, metastatic cancer, the postsplenectomy state, or iron deficiency anemia. Causes of primary thrombocytosis include myeloproliferative neoplasms, myelodysplastic syndromes, myelodysplastic syndrome/myeloproliferative neoplasm overlap syndromes, and other myeloid malignancies. Congenital thrombocytosis is extremely rare and sometimes associated with germline mutations of thrombopoietin or its receptor (MPL).
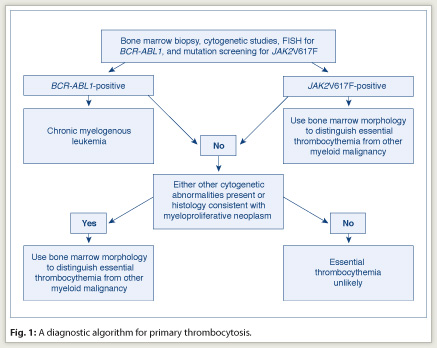
In managing the patient with thrombocytosis, one must first distinguish reactive from primary thrombocytosis. The presence of acute or subacute infection, a connective tissue disorder, vasculitis, hemolysis, active bleeding, recent surgery, history of splenectomy, or iron deficiency anemia favors the diagnosis of reactive thrombocytosis. The presence of chronic thrombocytosis, thrombohemorrhagic complications, microvascular symptoms, or splenomegaly favors the diagnosis of primary thrombocytosis. However, clinical impression often requires confirmation through laboratory testing, which is also necessary to distinguish among the different causes of primary thrombocytosis (including essential thrombocythemia) (see diagnostic workup in Fig. 1).
Below, I provide a stepwise approach to the laboratory evaluation of thrombocytosis.
 Step 1: Review the complete blood count and order blood smear examination
Step 1: Review the complete blood count and order blood smear examination
Complete blood count or blood smear clues for reactive thrombocytosis include presence of microcytic anemia (iron deficiency) or Howell-Jolly bodies (postsplenectomy state), which suggest thrombocytosis associated with surgical or functional hyposplenism. An increase in hematocrit or leukocyte count suggests myeloproliferative neoplasms, whereas presence of macrocytosis or leukoerythroblastic blood smear, or abnormal platelet morphology is consistent with clonal thrombocytosis. Microcytosis suggests iron deficiency anemia.
Step 2: Check serum ferritin and C-reactive protein
The initial laboratory tests should include the measurement of serum ferritin concentration. A normal serum ferritin level excludes the possibility of iron deficiency anemia–associated reactive thrombocytosis. However, a low level does not exclude the possibility of clonal thrombocytosis. Increased C-reactive protein suggests but does not secure a diagnosis of reactive thrombocytosis associated with an occult inflammatory or malignant process.
Step 3: Screen for JAK2V617F
JAK2V617F mutation screening is now part of the diagnostic workup for thrombocytosis, according to the World Health Organization (WHO) diagnostic criteria for essential thrombocythemia and polycythemia vera. However, peripheral blood mutation screening for JAK2V617F cannot substitute for bone marrow examination, since the mutation is not always detected in patients with essential thrombocythemia, where mutational frequency is estimated at 60%.
Step 4: Bone marrow examination
Bone marrow morphology appears normal in reactive thrombocytosis. In clonal thrombocythemia, bone marrow findings include increased numbers of megakaryocytes and other myeloid cells, abnormality in cell morphology, presence of megakaryocyte clusters, and reticulin fibrosis.
 In addition to morphologic analysis, bone marrow examination allows the performance of cytogenetic studies and special immunohistochemical stains. For example, the detection of a clonal cytogenetic abnormality is diagnostic of primary thrombocytosis. However, although some causes of primary thrombocytosis are always associated with an abnormal cytogenetic lesion (for example, chronic myelogenous leukemia [CML]), less than 5% of patients with essential thrombocythemia have detectable cytogenetic abnormalities.
In addition to morphologic analysis, bone marrow examination allows the performance of cytogenetic studies and special immunohistochemical stains. For example, the detection of a clonal cytogenetic abnormality is diagnostic of primary thrombocytosis. However, although some causes of primary thrombocytosis are always associated with an abnormal cytogenetic lesion (for example, chronic myelogenous leukemia [CML]), less than 5% of patients with essential thrombocythemia have detectable cytogenetic abnormalities.
Essential thrombocythemia is currently defined as a persistent thrombocythemic state (platelet count ≥ 450 × 109/L) that is neither reactive nor associated with an otherwise defined myeloid malignancy (including polycythemia vera), primary myelofibrosis (including prefibrotic primary myelofibrosis), CML, and myelodysplastic syndromes. In this regard, it should be noted that CML, myelodysplastic syndromes, and prefibrotic primary myelofibrosis can all mimic essential thrombocythemia in their presentation.
Therefore, before making a working diagnosis of essential thrombocythemia, in the context of primary thrombocytosis, one must exclude CML not only by conventional cytogenetics but also by a fluorescence in situ hybridization (FISH) test for BCR/ABL1. Similarly, bone marrow histology should be carefully scrutinized for the presence of both trilineage dysplasia—which would suggest myelodysplastic syndromes—and intense marrow cellularity accompanied by atypical megakaryocytic hyperplasia—which would suggest prefibrotic primary myelofibrosis.
Unlike essential thrombocythemia, prefibrotic primary myelofibrosis is often accompanied by elevated levels of serum lactate dehydrogenase level, increased peripheral blood CD34 cell count, and a leukoerythroblastic peripheral blood smear. The clinical relevance of distinguishing essential thrombocythemia from prefibrotic primary myelofibrosis has reently been demonstrated, where the latter was shown to be associated with significantly worse overall and leukemia-free survival.
Molecular Pathogenesis
The first evidence of monoclonality in essential thrombocythemia was suggested in the early 1980s by clonality studies that utilized glucose-6-phosphate dehydrogenase (G6PD) isoenzyme analysis. These early observations were subsequently confirmed by X-linked DNA analysis. Unlike the situation for CML, the primary molecular abnormality in essential thrombocythemia has not been identified. In this regard, cytogenetic studies have been of limited value, because they are conducted in less than 5% of patients at diagnosis and are nonspecific. Both structural and numerical abnormalities involving many individual chromosomes (including trisomies 9 and 8, and long-arm deletions of chromosomes 5, 7, 13, 17, and 20) have been associated with essential thrombocythemia.
Summary
The first step in approaching a patient with thrombocytosis is to entertain the possibility of reactive thrombocytosis, a process that has already been elaborated in a previous section. It is worth reiterating the value of mutation screening for JAK2V617F in distinguishing essential thrombocythemia from reactive thrombocytosis but not from other myeloproliferative neoplasms. In addition, rare cases of genetically defined essential thrombocythemia (eg, activating mutation of MPL) have been described and must be kept in mind while evaluating a patient with either a lifelong or family history of thrombocytosis.
The second step in evaluating a patient with thrombocytosis is to confirm the diagnosis of essential thrombocythemia with a bone marrow examination and exclude the possibility of other myeloid disorders, including prefibrotic primary myelofibrosis. Although detailed analysis of megakaryocyte morphology might assist in distinguishing CML (dwarf megakaryocytes and not too many clusters) from essential thrombocythemia (giant megakaryocytes with cluster formation), cytogenetic studies or FISH for BCR/ABL1 should accompany bone marrow examination to rule out the possibility of CML.
Mild reticulin fibrosis (grades 1 or 2) is detected in approximately 14% of patients with essential thrombocythemia at diagnosis and does not portend an unusual outcome. Clonal cytogenetic lesions in essential thrombocythemia are detected in < 5% of cases and are nonspecific. ■
Disclosure: Dr. Tefferi reported no potential conflicts of interest.
Suggested Readings
Barbui T, Thiele J, Passamonti F, et al: Survival and disease progression in essential thrombocythemia are significantly influenced by accurate morphologic diagnosis: An international study. J Clin Oncol 29:3179-3184, 2011.
Tefferi A: Polycythemia vera and essential thrombocythemia: 2012 update on diagnosis, risk stratification, and management. Am J Hematol 87:284-293, 2012.