
With In the Clinic, The ASCO Post provides overviews of novel oncology agents, addressing indications, mechanisms, administration recommendations, safety profiles, and other essential information needed for the appropriate clinical use of these drugs.
Sunitinib
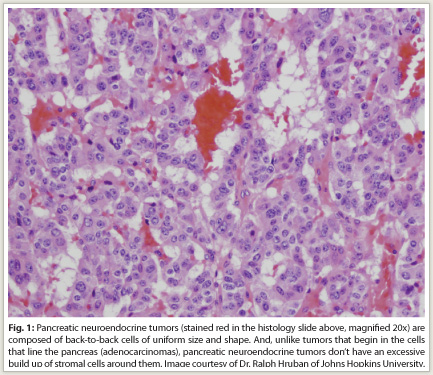 Indication—The oral kinase inhibitor sunitinib (Sutent) was recently granted approval for the treatment of progressive, well-differentiated pancreatic neuroendocrine tumors (pNET, Fig. 1) in patients with unresectable, locally advanced or metastatic disease. Sunitinib has earlier indications in gastrointestinal stromal tumors (GIST) after disease progression on imatinib (Gleevec) or in patients intolerant of imatinib and in advanced renal cell carcinoma.
Indication—The oral kinase inhibitor sunitinib (Sutent) was recently granted approval for the treatment of progressive, well-differentiated pancreatic neuroendocrine tumors (pNET, Fig. 1) in patients with unresectable, locally advanced or metastatic disease. Sunitinib has earlier indications in gastrointestinal stromal tumors (GIST) after disease progression on imatinib (Gleevec) or in patients intolerant of imatinib and in advanced renal cell carcinoma.
Approval of sunitinib in pNET is based on a randomized trial comparing sunitinib at 37.5 mg/d (n = 86) vs placebo (n = 85) in patients with unresectable, locally advanced or metastatic well-differentiated pNET. Treatment with somatostatin analogs was allowed. A total of 69% of patients given placebo received sunitinib after progression. The trial was discontinued early, after the independent data and safety monitoring committee observed more serious adverse events and deaths in the placebo group, as well as a difference in progression-free survival favoring sunitinib.
Sunitinib treatment significantly prolonged median progression-free survival, the trial’s primary endpoint, from 5.4 to 10.2 months, representing a 57% reduction in risk of progression (hazard ratio = 0.43, P < .001). Improved progression-free survival was seen in multiple patient subgroups, including those with prior somatostatin analog treatment. Overall survival data were not mature at the time of analysis. The overall response rate with sunitinib was 9.3% (no complete responses).
 How it works—Sunitinib inhibits cellular signaling by targeting multiple receptor tyrosine kinases. Vascular endothelial growth factor (VEGF) is a key driver of angiogenesis in pancreatic neuroendocrine tumors, and malignant pancreatic endocrine tumors also show widespread expression of platelet-derived growth factor receptors (PDGFRs) α and β, stem-cell factor receptor (c-kit), and VEGF receptor (VEGFR)-2 and VEGFR-3. Sunitinib inhibits these kinases, and has been shown to delay tumor growth in a RIP1-Tag2 transgenic mouse model of pancreatic islet cell tumors by reducing endothelial cell density and pericyte coverage of tumor vessels.
How it works—Sunitinib inhibits cellular signaling by targeting multiple receptor tyrosine kinases. Vascular endothelial growth factor (VEGF) is a key driver of angiogenesis in pancreatic neuroendocrine tumors, and malignant pancreatic endocrine tumors also show widespread expression of platelet-derived growth factor receptors (PDGFRs) α and β, stem-cell factor receptor (c-kit), and VEGF receptor (VEGFR)-2 and VEGFR-3. Sunitinib inhibits these kinases, and has been shown to delay tumor growth in a RIP1-Tag2 transgenic mouse model of pancreatic islet cell tumors by reducing endothelial cell density and pericyte coverage of tumor vessels.
How it is given—The recommended dose of sunitinib for pNET is 37.5 mg taken orally once daily continuously without a scheduled off-treatment period. Sunitinib can be taken with or without food. Patients with severe or intolerable adverse reactions may require temporary dose reductions to 25 mg/d or dose interruption.
Safety profile—Sunitinib has a boxed warning for hepatotoxicity. During the double-blind phase of the trial supporting approval of sunitinib in pNET, the most common (≥ 30%) adverse reactions were diarrhea, nausea, asthenia, vomiting, and fatigue. The most common (≥ 5%) grade 3/4 adverse reactions were neutropenia, hypertension, palmar-plantar erythrodysesthesia syndrome, and leukopenia. Five patients receiving sunitinib and nine receiving placebo died on-study; of the five sunitinib on-study deaths, one was due to cardiac failure and four were due to disease progression. An additional sunitinib patient was taken off study due to cardiac failure and died 2 months later.
Adverse events resulted in permanent discontinuation in 22% of sunitinib patients and in 17% of placebo patients. Dose delays or reductions were required in 61% and 23% of patients, respectively.
Suggested Readings
Kulke MH, Lenz HJ, Meropol NJ, et al: Activity of sunitinib in patients with advanced neuroendocrine tumors. J Clin Oncol 26:3403-3410, 2008.
Raymond E, Dahan L, Raoul JL, et al: Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. N Engl J Med 364:501-513, 2011.
SUTENT® (crizotinib capsules, oral) prescribing information. Pfizer Labs, May 2011. Available at http://labeling.pfizer.com/ShowLabeling.aspx?id=607. Accessed October 24, 2011.
U.S. Food and Drug Administration: What’s New from the Office of Hematology Oncology Products: Sunitinib. Available at http://www.fda.gov/AboutFDA/CentersOffices/CDER/ucm256499.htm. Accessed October 24, 2011.
Everolimus
 Indication—The kinase inhibitor everolimus (Afinitor) was also recently approved for treatment of unresectable, locally advanced or metastatic progressive neuroendocrine tumors of pancreatic origin (pNET). The safety and efficacy of everolimus in treatment of carcinoid tumors have not been established. Everolimus has prior indications in advanced renal cell carcinoma after failure of sunitinib or sorafenib and in subependymal giant cell astrocytoma associated with tuberous sclerosis that requires intervention but cannot be treated by curative resection.
Indication—The kinase inhibitor everolimus (Afinitor) was also recently approved for treatment of unresectable, locally advanced or metastatic progressive neuroendocrine tumors of pancreatic origin (pNET). The safety and efficacy of everolimus in treatment of carcinoid tumors have not been established. Everolimus has prior indications in advanced renal cell carcinoma after failure of sunitinib or sorafenib and in subependymal giant cell astrocytoma associated with tuberous sclerosis that requires intervention but cannot be treated by curative resection.
Approval was based on a randomized trial of everolimus 10 mg/d (n = 207) vs placebo (n = 203) in patients with unresectable, locally advanced or metastatic pNET. Treatment with somatostatin analogs was allowed as part of best supportive care. In total, 73% of placebo patients crossed over to everolimus after disease progression, as permitted by study protocol. Median progression-free survival, the primary endpoint of the trial, was increased from 4.6 to 11.0 months with everolimus, representing a 65% reduction in risk of progression (HR = 0.35, P < .001). Improvements in progression-free survival were observed across all patient subgroups, irrespective of prior somatostatin analog use. An interim analysis showed no difference between groups in overall survival (HR = 1.05, P = NS). The response rate with everolimus was 4.8%, with no complete responses.
 How it works—Everolimus inhibits mammalian target of rapamycin (mTOR), a serine-threonine kinase that stimulates cell growth, proliferation, and angiogenesis. Autocrine activation of the mTOR signaling pathway, which is mediated by insulin-like growth factor 1, is implicated in the proliferation of pancreatic neuroendocrine tumor cells, and inhibition of mTOR has been shown to result in an antiproliferative effect.
How it works—Everolimus inhibits mammalian target of rapamycin (mTOR), a serine-threonine kinase that stimulates cell growth, proliferation, and angiogenesis. Autocrine activation of the mTOR signaling pathway, which is mediated by insulin-like growth factor 1, is implicated in the proliferation of pancreatic neuroendocrine tumor cells, and inhibition of mTOR has been shown to result in an antiproliferative effect.
How it is given—The recommended dose of everolimus is 10 mg once daily; it should be taken at the same time of day and consistently with or without food. Patients with severe or intolerable adverse reactions may require temporary dose reductions to 5 mg/d or dose interruption. The dose should be reduced to 5 mg once daily in patients with moderate (Child-Pugh class B) hepatic impairment. The starting dose should be 2.5 mg once daily if treatment with drugs that are moderate inhibitors of CYP3A4 and/or P-glycoprotein is required; if tolerated, the dose can be increased to 5 mg once daily. If strong inducers of CYP3A4 are required, the dose can be increased in 5-mg increments to a maximum of 20 mg once daily.
Safety profile—Data from the trial supporting everolimus approval and 858 additional patients with advanced neuroendocrine tumors in the everolimus safety database indicate that the most common adverse reactions in patients receiving everolimus (> 30%) were stomatitis, rash, diarrhea, fatigue, edema, abdominal pain, nausea, fever, and headache. The most common (≥ 5%) grade 3 or 4 adverse reactions were stomatitis and diarrhea. The most common (≥ 3%) grade 3 or 4 laboratory abnormalities were hyperglycemia, lymphopenia, decreased  hemoglobin, hypophosphatemia, increased ALP, neutropenia, increased AST, decreased potassium, and thrombocytopenia.
hemoglobin, hypophosphatemia, increased ALP, neutropenia, increased AST, decreased potassium, and thrombocytopenia.
During the trial, death occurred in 7 everolimus patients and 1 placebo patient. Causes of death in the everolimus group included acute renal failure, acute respiratory distress, cardiac arrest, death (cause unknown), hepatic failure, pneumonia, sepsis, and death from unknown cause. After crossover to open-label everolimus, there were 3 additional deaths, 1 due to hypoglycemia and cardiac arrest in a patient with insulinoma, 1 due to myocardial infarction with congestive heart failure, and 1 due to sudden death. Adverse events resulted in permanent discontinuation in 20% and 6% of patients in the everolimus and placebo groups, respectively. Dose delays or reductions were necessary in 61% of everolimus patients and 29% of placebo patients. Opportunistic infections in patients with advanced neuroendocrine tumors included hepatitis B reactivation (resulting in death), mycobacterial infection, and invasive aspergillus. Pneumonitis was seen in 11% of patients (grade 3 or 4 in 1.7%) in the safety database. ■
Suggested Readings
AFINITOR® (everolimus) tablets prescribing information. Novartis Pharmaceuticals Corporation, July 2011. Available at http://www.pharma.us.novartis.com/product/pi/pdf/afinitor.pdf. Accessed October 24, 2011.
Missiaglia E, Dalai I, Barbi S, et al: Pancreatic endocrine tumors: Expression profiling evidences a role for AKT-mTOR pathway. J Clin Oncol 28:245-255, 2010.
U.S. Food and Drug Administration: What’s New from the Office of Hematology Oncology Products: Everolimus. Available at http://www.fda.gov/AboutFDA/CentersOffices/CDER/ucm254392.htm. Accessed October 24, 2011.
von Wichert G, Jehle PM, Hoeflich A, et al: Insulin-like growth factor-I is an autocrine regulator of chromogranin A secretion and growth in human neuroendocrine tumor cells. Cancer Res 60:4573-4581, 2000.
Yao JC, Lombard-Bohas C, Baudin E, et al: Daily oral everolimus activity in patients with metastatic pancreatic neuroendocrine tumors after failure of cytotoxic chemotherapy: A phase II trial. J Clin Oncol 28:69-76, 2010.
Yao JC, Shah MH, Ito T, et al: Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med 364:514-523, 2011.